
Abstract
Accrual of ceramides, membrane and bioactive sphingolipids, has been implicated in endothelial dysfunction preceding cardiometabolic diseases. Yet, direct in vivo evidence, underlying mechanisms, and pathological implications are lacking. Here we show that suppression of ceramides and sphingosine-1-phosphate (S1P), a product of ceramide degradation, are causally linked to endothelial dysfunction and activation, contributing to vascular and metabolic disorders in high fat diet fed (HFD) male mice. Mechanistically, the upregulation of Nogo-B and ORMDL proteins suppress ceramide de novo biosynthesis in endothelial cells (EC) of HFD mice, resulting in vascular and metabolic dysfunctions. Systemic and endothelial specific deletion of Nogo-B restore sphingolipid signaling and functions, lowers hypertension, and hepatic glucose production in HFD. Our results demonstrate in vivo that ceramide and S1P suppression rather than accrual contributes to endothelial dysfunction and cardiometabolic diseases in HFD mice. Our study also sets a framework for the development of therapeutic strategies to treat these conditions
Similar content being viewed by others
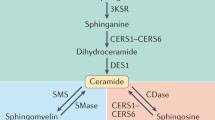


Introduction
The incidence of cardiometabolic diseases is growing at alarming rates. Recent data show that >1.6 billion of people worldwide are overweight or obese, a condition causally linked to diabetes, non-alcoholic liver steatosis and cardiovascular (CV) diseases1.
Endothelial dysfunction is an early event in the onset of cardiometabolic diseases, including diabetes, obesity, and atherosclerosis2,3,4, and emerging studies have shown that can also enhance metabolic dysregulations5.
Ceramide and sphingosine-1-phosphate (S1P) are major bioactive sphingolipids regulating a variety of cellular processes including proliferation, migration, and endothelial functions relevant in CV health and diseases4,6,7,8. Postnatally, S1P signaling decreases vascular tone and BP by activating eNOS mainly via S1P receptor 1(S1P1)9, the most abundant S1P receptor in EC. The endothelium is also a source of S1P4,8, the production of which is stimulated by flow10 and can induce NO-mediated vasorelaxation8,9,11. Single nucleotide polymorphisms (SNP) in genes of S1P pathway, including S1pr112, Sphk113, and Rtn414, have been linked to atherosclerosis suggesting that S1P signaling is important to preserve CV functions.
As membrane components, ceramides can influence the biophysical properties of cell membranes and lipid rafts15,16, important signaling platforms. Genetic suppression of ceramide de novo biosynthesis in EC, by deleting Sptlc2, a subunit of serine palmitoyltransferase (SPT), first and rate limiting enzyme of the pathway, impaired signal transduction, flow-induced vasodilation and blood pressure (BP) regulation, suggesting that ceramide homeostasis is necessary to preserve endothelial functions16. Ceramides are also second messengers and can modulate the activity of target proteins, such as protein phosphatases PP2A and PP117,18. However, how ceramide metabolism changes and contributes to endothelial dysfunction and cardiometabolic diseases in vivo remain an open question.
In vitro studies showed that high glucose and/or palmitate, mimicking the in vivo conditions of diabetes and/or metabolic syndrome, boosted ceramide production19,20,21, which induced ROS formation22, protein phosphate 2A (PP2A) activation and eNOS dephosphorylation at Ser11176, all resulting in decreased nitric oxide (NO) production and bioavailability4,19,20. Whether the accrual of ceramides occurs in vivo and contributes to endothelial dysfunction in cardiometabolic disorders have yet to be demonstrated. This is especially important when aiming to translate preclinical data to the clinic.
Furthermore, ceramides were reported elevated in the thoracic aorta of obese diabetic mice19, hence it was inferred that similar metabolic changes occurred also in resistance arteries. However, capacitance and resistance arteries are structurally and functionally different, and whether ceramides accumulate in resistance arteries and contribute to dysfunction and hypertension in cardiometabolic disorders remains an open question.
We discovered that in the endothelium of HFD-fed mice, ceramide de novo biosynthesis is suppressed, resulting in endothelial dysfunction and activation of pro-inflammatory gene expression. These changes underlie both vascular and metabolic dysfunction. Mechanistically, the upregulation of ORMDL23 and Nogo-B11 proteins, inhibitors of SPT activity, suppresses sphingolipid de novo biosynthesis in EC. Interestingly, systemic, and endothelial deletion of Nogo-B restored endothelial sphingolipid levels and signaling in HFD mice, hence improving vascular functions and hypertension. Furthermore, endothelial Nogo-B deletion also refrained hepatic gluconeogenesis, in part via S1P, thus ameliorating diabetes.
Our data revealed that in HFD conditions ceramide de novo biosynthesis is suppressed in EC and underlies both vascular and metabolic dysfunctions of diabetes. The findings of this study, the first to investigate local mechanisms of ceramide metabolism derangement and its pathological implications, are paradigm shift for the long-standing believe that endothelial ceramide accrual underlies cardiometabolic diseases.
Results
Ceramide de novo biosynthesis is suppressed in both EC and resistance arteries of obese and diabetic mice and correlates with endothelial pro-inflammatory gene expression
To directly measure how HFD impacts ceramide metabolism and levels in EC in vivo, C57BL/6 J WT mice were fed with standard diet (SD) or HFD for 6 months (Fig. 1A). As expected, HFD feeding significantly increased body weight (Fig. 1B) and induced glucose intolerance compared to SD-fed mice (Fig. 1C). After 6 mo of SD or HFD, EC from heart and lung were FACS-sorted and the expression of the SPT complex was assessed via WB analysis. Interestingly, both NOGO-B and ORMDLs, inhibitors of SPT activity, were significantly upregulated in EC from HFD mice, whereas SPTLC1 and SPTLC2 were unchanged (Fig. 1D–F). RNAseq analysis of FACS-sorted EC showed a significant upregulation of Rtn4 (encoding Nogo proteins), Ormdl3 and Ormdl2 genes in HFD in line with WB data (Fig. 1G). Furthermore, cytokines (i.e. Il1a, Il1b), adhesion molecules (i.e. Icam1, Vcam1, Sele), and atherosclerotic genes (i.e. increase in Ldlr and decrease in abca1, abcg1) were markedly elevated, indicating a pro-inflammatory phenotype of the EC in HFD (Fig. 1G).

A Schematic representation of the experimental design and of the sphingolipid pathway. Male WT mice were fed with SD or HFD for 6 months (Created in BioRender. Di Lorenzo, A. (2025)). B Body weight (WT SD n = 10 mice and WT HFD n = 9 mice). C Glucose tolerance test (GTT) (WT SD n = 15 mice, WT HFD n = 9 mice). D WB analysis of NOGO-B, ORMDLs, SPTLC1 and SPTLC2 in EC FACS-sorted from heart and lung and E, F relative quantification (WT SD n = 7 mice, WT HFD n = 8 mice). β-ACTIN was used as loading control. G Heatmap of genes for the SPT complex, inflammation, and lipid transport in FACS-sorted EC from heart and lung, *indicates that the difference is significant (WT SD n = 4 mice and WT HFD n = 4 mice). LC–MS/MS quantification of H total ceramides, I specific ceramides, C16:0-dihydroceramide (dhC16:0-Cer), dihydrosphingosine (dhSph), sphingosine (Sph), sphingosine-1-phosphate (S1P), and J total and K specific sphingomyelins (SM) in FACS-sorted EC from heart and lung (WT SD n = 16 mice, WT HFD n = 18 mice). L Immunofluorescence staining for NOGO-A/B and WGA of mesenteric arteries (MA) sections. Nogo-A/B deficient MA were used as negative control. Nuclei were stained with DAPI. LC–MS/MS quantification of M total and N specific Cer, and O total and P specific SM in MA (WT SD n = 9 mice and WT HFD n = 11 mice). Data are expressed as mean ± SEM *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. Statistical significance was determined by unpaired t-test two-tailed (B, E, F, H–K, M–P) and two-way ANOVA with Sidak multiple comparison test (C). Source data are provided as a Source Data file.
Consistently, ceramides, S1P (Fig. 1H, I) and sphingomyelins (SM, complex sphingolipids derived from ceramides Fig. 1J, K) were markedly decreased in EC of HFD- versus SD-fed mice. Obesity and diabetes are known to set off a pro-inflammatory phenotype of the vasculature24,25. These data suggest that the suppression rather than accrual of ceramides correlates with the endothelial activation in HFD conditions.
Next, we assessed NOGO-B expression and sphingolipid profiles in mesenteric arteries (MA) of obese and lean mice. Immunofluorescence staining revealed a remarkable upregulation of NOGO-B in the vascular wall of HFD vs. SD arteries (Fig. 1L). Consistently, ceramides (Fig. 1M, N) and sphingomyelins (Fig. 1O, P) were significantly decreased in MA of obese mice, suggesting that the downregulation rather than the upregulation of this pathway is associated with the endothelial dysfunction and hypercontractility of resistance arteries in HFD conditions.
Deletion of Nogo-A/B reduces hepatic glucose production without affecting body weight in lean mice
To investigate the pathological significance of Nogo-B upregulation in EC and resistance arteries we used Nogo-A/B-deficient mice (Fig. 2A). First, we assessed the metabolic phenotype of Nogo-A/B-deficient mice and their controls (WT littermates). Body weight, fat, and lean mass contents were not different in Nogo-A/B-deficient mice and age-matched controls on SD up to 6 months (Fig. 2B, C). Food intake (Fig. 2D), energy expenditure (Supplementary Fig. 1A, B), respiratory quotients (Supplementary Fig. 1C–E), water intake (Supplementary Fig. 1F, G) and locomotor activity (Supplementary Fig. 1H), were not different between the two groups. Nogo-A/B-deficient mice showed a significant reduction in fasting plasma glucose (Fig. 2E), improved glucose tolerance (Fig. 2F), with no change in insulin sensitivity (Fig. 2G) and plasma insulin levels (Fig. 2H), although Nogo-A/B proteins are expressed in the β-cells of pancreatic islets26 (Fig. 2I). Interestingly, Nogo-A/B-deficient mice showed a remarkable suppression of hepatic glucose production compared to controls as shown by the pyruvate tolerance test (PTT, Fig. 2J), in line with a significant lower expression of G6Pase (Fig. 2K). Conversely, the acute treatment of the mice with myriocin, an inhibitor of SPT, induced a significant upregulation of glucose production as shown by the PTT (Fig. 2L), suggesting a role for de novo produced sphingolipids in hepatic glucose metabolism. In the liver, Nogo-A/B proteins are expressed in the vasculature, epithelial cells of the bile ducts, and stellate cells but not in hepatocytes27 (Fig. 2M), suggesting that modulating SPT activity in these cell types, but not in hepatocytes, might impact gluconeogenesis via paracrine mechanisms. We reported that mice lacking Nogo-B have increased plasma S1P11 and different studies implicated a role for S1P synthetizing enzymes, sphingosine kinase 1 and 2 (SPHK1 and SPHK2), in regulating hepatic glucose metabolism. Sphk1 gene delivery significantly reduced the blood glucose level of in a mouse model of T2D28, and liver specific deletion of Sphk2 was recently shown to enhance the hepatic glucose production29. Considering that hepatocytes do not express Nogo-B, we hypothesized that local S1P (i.e. endothelial-derived) impacts hepatic gluconeogenesis. By using primary murine hepatocytes in culture, we demonstrated that exogenous S1P significantly reduced glucose production, an effect that was abolished by W146, an inhibitor of S1P1 (Fig. 2N), suggesting that S1P downregulates hepatocyte glucose production via S1P1 signaling. Interestingly, the administration of W146 in SD WT mice was sufficient to markedly elevate plasma glucose levels (Fig. 2O, right vs. left graph), supporting the role of S1P1 signaling in glucose homeostasis.

A Schematic representation of the experimental design. All the analysis in this figure were conducted on male WT and Nogo-A/B-deficient mice SD-fed for 6 months (Created in BioRender. Di Lorenzo, A. (2025) https://BioRender.com/i50g226). B Body weight (n = 18 mice per group), C Fat mass, lean mass and total water content (n = 5 mice per group). D Total, dark phase, and light phase food intake, expressed as average of 3 consecutive days (n = 5 mice per group). E Plasma glucose levels after o.n. starvation (WT SD n = 14 mice and Nogo-A/B-deficient SD n = 9 mice). F GTT (WT SD n = 19 mice and Nogo-A/B-deficient n = 13 mice) and G ITT (n = 9 mice per group). H Plasma insulin levels pre- and post-glucose administration (i.p., 1.5 g/kg). (WT SD n = 8 mice and Nogo-A/B-deficient SD n = 7 mice). I Immunofluorescence staining for glucagon, insulin and NOGO-A/B in pancreatic sections of SD WT mice. Nogo-A/B-deficient pancreas were used as negative controls. J PTT (WT SD n = 15 mice and Nogo-A/B-deficient SD n = 8 mice) and K G6Pase mRNA levels in the liver (n = 5 mice per group). L PTT in WT and Nogo-A/B-deficient mice treated with vehicle or Myriocin (0.3 mg/kg, o.n.) (WT SD vehicle n = 8 mice, WT SD treated with myriocin n = 10, Nogo-A/B-deficient SD vehicle n = 8 mice and Nogo-A/B-deficient SD treated with myriocin n = 10). M Immunohistochemistry for NOGO-A/B in liver of lean mice. Nogo-A/B deficient liver were used as negative control. N Glucose production from hepatocytes isolated from WT mice, treated with vehicle or S1P (1 μM, 6 h), and/or W146 (1 μM, 6 h) (n = 3 independent experiments). O PTT in WT male mice on SD after 30 min from intraperitoneal administration of W146 (3 mg/kg) or vehicle (DMSO:water, 1:1) after o.n. starvation (WT SD vehicle n = 6 mice, WT SD treated with W146 n = 8, Nogo-A/B-deficient SD vehicle n = 5 mice and Nogo-A/B-deficient SD treated with W146 n = 7). Data were expressed as mean ± SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. Statistical analysis was performed with unpaired t-test two-tailed (B–E, K), one-way ANOVA (N) and two-way ANOVA with Sidak multiple comparisons test (F, G, H, J, L, O). Source data are provided as a Source Data file.
Deletion of Nogo-A/B refrains obesity and hepatic glucose production
Nogo-A/B-deficient mice and their controls were fed with HFD for 6 months (Fig. 3A). Body weight was significantly lower in Nogo-A/B-deficient vs. controls (Fig. 3B) and corresponded to a significantly reduced fat mass (Fig. 3C), as well as leptin levels (Fig. 3D). The differences in body weight and composition were associated with a significant reduction in food intake observed specifically during the dark phase (Fig. 3E). Differences were not associated to changes in energy expenditure (Supplementary Fig. 2A, B), O2 consumption, CO2 production or respiratory exchange ratio (Supplementary Fig. 2C–E), and water intake (Supplementary Fig. 2F, G). Locomotor activity was also no different between the two groups (Supplementary Fig. 2H). Nogo-A/B-deficient mice showed a significant reduction in fasting plasma glucose (Fig. 3F) and improved glucose tolerance (Fig. 3G), with no difference in insulin sensitivity (Fig. 3H) and in plasma insulin levels, both in fasting and after glucose administration (Fig. 3I). Hepatic glucose production was significantly reduced in Nogo-A/B-deficient compared to control mice (Fig. 3 J). However, this effect was not due to the expression of Nogo-B in hepatocytes (Fig. 3K). Finally, no significant differences were observed in liver weight (Fig. 3L), triglyceride (Fig. 3M), and DAG content (Supplementary Fig. 3A). On the contrary, ceramides, hexosylceramides and sphingomyelins were significantly reduced in HFD Nogo-A/B-deficient mice (Supplementary Fig. 3B–D), which could also contribute to the lower hepatic glucose production30.

A Schematic representation of the experimental design. All the analysis were performed in male WT and Nogo-A/B-deficient mice HFD-fed for 6 months (Created in BioRender. Di Lorenzo, A. (2025) https://BioRender.com/i50g226). B Body weight (n = 19 mice per group). C Fat mass, lean mass, and total water content (n = 5 mouse per group). D Plasma leptin levels (WT HFD n = 9 mice and Nogo-A/B-deficient HFD n = 10 mice). E Total, dark phase, and light phase food intake, expressed as average of 3 consecutive days (n = 5 mice per group). F Plasma glucose levels after o.n. starvation (WT HFD n = 16 mice and Nogo-A/B-deficient HFD n = 20 mice). G GTT (WT HFD n = 25 mice and Nogo-A/B-deficient HFD n = 19 mice). H ITT (WT HFD n = 27 mice and Nogo-A/B-deficient HFD n = 26 mice). I Plasma insulin levels pre- and post-glucose administration (i.p.; 1.5 g/kg) (WT HFD n = 17 mice and Nogo-A/B-deficient HFD n = 25 mice). J PTT (WT HFD n = 16 mice and Nogo-A/B-deficient HFD n = 15 mice). K Immunohistochemistry for NOGO-A/B in liver of WT mice HFD-fed for 6 months. Nogo-A/B deficient liver were used as negative control. L Liver weight (n = 19 mice per group). M Triglyceride levels (n = 5 mice per group). Data were expressed as mean ± SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. Statistical analysis was performed with unpaired t-test two-tailed (C, D, E, F, L, M), one-way ANOVA (I), and two-way ANOVA with Sidak multiple comparisons test (B, G, H, J). Source data are provided as a Source Data file.
The loss of Nogo-A/B lowers blood pressure in both lean and obese diabetic mice
Radiotelemetry measurements (Fig. 4A) showed a significant decrease in systolic (SBP), diastolic (DBP), and mean BP (MBP) in lean Nogo-A/B-deficient mice vs. control (Fig. 4B–D, first column), consistent with previous data obtained by tail cuff system on younger mice11. SBP was significantly increased in obese WT and Nogo-A/B-deficient mice compared to their lean controls (Fig. 4B–D, second and third column). However, in obese Nogo-A/B-deficient mice SBP was significantly lower vs. obese WT (Fig. 4B–D, last column). No differences were observed in the heart rate (Supplementary Fig. 4). These data suggest that suppression of sphingolipid metabolism and signaling by Nogo-B contribute to hypertension in obesity and diabetes.

A Radiotelemetry measurements of blood pressure (BP) (Created in BioRender. Di Lorenzo, A. (2025) https://BioRender.com/i50g226). Systolic BP (SBP; B), diastolic BP (DBP; C), and mean BP (MBP; D) were measured by radiotelemetry for 3 consecutive days following the recovery from surgery of ca. 12 days. (Nogo-A/B-deficient SD, n = 6 mice; Nogo-A/B-deficient HFD, n = 9 mice, WT SD and HFD, n = 10 mice per group). Data were expressed as mean ± SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. Statistical analysis was performed with two-way ANOVA with Sidak multiple comparisons test. Source data are provided as a Source Data file.
Deletion of Nogo-A/B protects from vascular dysfunction by preserving sphingolipid metabolism and levels
To study the impact of Nogo-A/B deletion on vascular function, we assessed the vasomotor reactivity of MA ex vivo by using the pressure myograph system (Fig. 5A). While acetylcholine31-induced vasodilation was not different in SD-fed mice, it was significantly improved in mice Nogo-A/B deficient vs. WT on HFD for 6 months (Fig. 5B). S1P-S1P1 signaling is a key mediator of flow-induced vasodilation8,9. The deletion of Nogo-B in MA improved flow-mediated vasodilation in lean and obese mice compared to WT mice in the same conditions (Fig. 5C, left). This difference was abolished by W146, an S1P1 inhibitor (Fig. 5C, right), suggesting that in absence of Nogo-B, local S1P signaling in response to flow was enhanced. Heighten vascular contraction and arterial remodeling contribute to the pathophysiology of hypertension32. Vasoconstriction in response to phenylephrine (PE, Fig. 5D, E) myogenic tone (Fig. 5F), and angiotensin-II (AngII, Fig. 5G) were significantly reduced in Nogo-A/B-deficient mice vs. WT in HFD.

A Vascular function of mesenteric arteries (MA) was assessed in WT or Nogo-A/B-deficient mice fed with SD or HFD for 6 months by using the pressure myograph system (Danish MyoTechnology, Aarhus, Denmark), (Created in BioRender. Di Lorenzo, A. (2025) https://BioRender.com/i50g226). B Ach-mediated vasodilation (WT SD n = 6 mice, Nogo-A/B-deficient SD n = 4 mice, WT HFD n = 6 mice, Nogo-A/B-deficient HFD n = 6 mice); C Flow-induced vasodilation (WT SD n = 6 mice, Nogo-A/B-deficient SD n = 5 mice, WT HFD n = 5 mice, Nogo-A/B-deficient HFD n = 6 mice). HFD MA of WT or Nogo-A/B-deficient mice were incubated with W146 (300 nM, 30 min), an S1P1 inhibitor (WT SD n = 6 mice, Nogo-A/B-deficient SD n = 5 mice, WT HFD n = 5 mice, Nogo-A/B-deficient HFD n = 6 mice, WT HFD n = 4 mice and Nogo-A/B-deficient HFD n = 3 mice treated with W146); D, E PE-induced vasoconstriction (WT SD n = 6 mice, Nogo-A/B-deficient SD n = 4 mice, WT HFD n = 6 mice, Nogo-A/B-deficient HFD n = 6 mice); F myogenic tone (WT SD n = 7 mice, Nogo-A/B-deficient SD n = 4 mice, WT HFD n = 6 mice and Nogo-A/B-deficient HFD n = 6 mice); and G AngII-induced vasoconstriction (WT SD n = 4 mice, Nogo-A/B-deficient SD n = 4 mice, WT HFD n = 4 mice and Nogo-A/B-deficient HFD n = 4 mice) in MA. LC–MS/MS quantification of H total and I specific Cer, and of J total and K specific SM in MA (WT HFD n = 11 mice, Nogo-A/B-deficient HFD n = 9 mice). Data are expressed relative to WT HFD. L WB analysis of p-MLC (Ser19), MLC, p-MYPT1 (Thr696), MYPT1 and NOGO-B in MA from obese WT or Nogo-A/B-deficient mice, with or without AngII stimulation (10−7M, 20 s) and M, N relative quantification (n = 4 mice per group). Data were expressed as mean ± SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. Statistical analysis was performed with two-way ANOVA with Sidak multiple comparisons test (B–G) and unpaired t-test two tailed (H–K, M, N). Source data are provided as a Source Data file.
In MA of HFD mice, smooth muscle cells, the bulk of the vascular wall, showed an upregulation of Nogo-B in line with a decrease of ceramides (Fig. 1I, L). Conversely, the loss of Nogo-B preserved ceramides levels in MA of HFD mice (Fig. 5 H, I), but not sphingomyelins (Fig. 5J, K), likely due to other regulatory mechanisms of sphingolipids involved in this setting and/or a less prevalent role of Nogo-B in smooth muscle cells compared to the endothelium.
Increased ceramides have been correlated with a reduction in MLC phosphorylation33, hence contractility. Thus, we performed WB analysis on MA for myosin light chain (MLC) and myosin phosphatase target subunit 1 (MYPT1), a subunit of MLC phosphatase34. No difference was observed in MA of lean mice in P-MLC following AngII stimulation (Supplementary Fig. 5), in line with similar contractility (Fig. 5G). Interestingly, while MLC and MYPT1 phosphorylation was significantly upregulated in MA of HFD-fed WT mice, in support of the enhanced vasocontractility (Fig. 5L–N), in Nogo-A/B-deficient MA were markedly diminished, suggesting that in obesity the upregulation of Nogo-B in smooth muscle cells contributes to the hypercontractility of the vasculature and hypertension by suppressing ceramide de novo production.
Endothelial-specific deletion of Nogo-A/B improves metabolic and vascular dysfunction
Nogo-B is the major isoform expressed in EC, while Nogo-A is abundant in the brain35 and in cardiomyocytes of hypertrophic hearts6. To dissect whether improved endothelial functions in absence of Nogo-A/B resulted from locally preserved ceramide and S1P signaling or from the improved metabolic milieu, we used endothelial-specific Nogo-A/B knockout mice (Nogo-A/BECKO, Fig. 6A)11. In SD there were no difference in body weight, GTT and ITT between the two groups (Supplementary Fig. 6). Body weight was not different in Nogo-A/BECKO mice and age-matched controls (Nogo-A/Bf/f) on HFD up to 6 months (Fig. 6B). HFD-fed Nogo-A/BECKO mice showed a significant improvement in glucose tolerance (Fig. 6C), with no difference in insulin sensitivity (Fig. 6D). Interestingly, PTT showed that hepatic glucose production was significantly reduced in obese Nogo-A/BECKO mice vs. WT (Fig. 6E), with no differences in glucagon (Supplementary Fig. 7). These data support a paracrine effect of the endothelium on the hepatocytes, in part attributable to S1P signaling, in agreement S1P inhibition of glucose production in hepatocytes (Fig. 2N). Radiotelemetry measurements showed a significant decrease in SBP (Fig. 6F–H) in Nogo-A/BECKO mice vs. WT fed with SD for 6 months. HFD significantly increased SBP and MBP in both Nogo-A/Bf/f and Nogo-A/BECKO mice, compared to their respective controls on SD (Fig. 6F–H). However, in HFD state, SBP was significantly lower in Nogo-A/BECKO compared to Nogo-A/Bf/f (Fig. 6F–H). To study the impact of endothelial Nogo-B deletion on vascular functions, we assessed the vasomotor reactivity of MA ex vivo. Vasoconstriction to PE and myogenic tone were significantly reduced in HFD Nogo-A/BECKO vs. Nogo-A/Bf/f MA (Fig. 6I–L). Ach-induced vasodilation was significantly improved in Nogo-A/BECKO MA from both SD and HFD mice, compared to control (Fig. 6M, N). The loss of endothelial Nogo-B significantly improved flow-mediated vasodilation in SD and HFD (Fig. 6O, P). Interestingly, myriocin, inhibitor of SPT, suppressed vasodilation to flow to the same extend in both genotypes, suggesting that the upregulation of sphingolipid de novo biosynthesis underlined the enhanced vasodilation in absence of Nogo-B. Interestingly, improved endothelial function of HFD Nogo-A/BECKO MA correlated with preserved ceramides and S1P (Fig. 6R, S) and sphingomyelins (Fig. 6T, U) in EC. Finally, RNAseq analysis on FACS-sorted EC from HFD Nogo-A/BECKO showed a significant downregulation in the expression of adhesion molecules (i.e. Sele, Selp) and lipid transporters (i.e. Ldlr, Lipg) compared to Nogo-A/Bf/f EC (Fig. 6V). These data suggest that the loss of Nogo-B preserves endothelial functions by maintaining vasculature sphingolipid homeostasis.

A Schematic representation of the experimental design. Metabolic and vascular functions were assessed in Nogo-A/Bf/f or Nogo-A/BECKO mice fed with SD or HFD for 6 months (Created in BioRender. Di Lorenzo, A. (2025) https://BioRender.com/i50g226). B Body weight (Nogo-A/Bf/f HFD n = 8 mice and Nogo-A/BECKO HFD n = 10 mice). C GTT (Nogo-A/Bf/f HFD n = 15 mice and Nogo-A/BECKO HFD n = 17 mice). D ITT (Nogo-A/Bf/f HFD n = 8 mice and Nogo-A/BECKO HFD n = 10 mice). E PTT (Nogo-A/Bf/f HFD n = 8 mice and Nogo-A/BECKO HFD n = 8 mice). F SBP, G DBP, and H MBP measured by radiotelemetry as described in Methods (Nogo-A/Bf/f SD n = 5 mice, Nogo-A/BECKO SD n = 5 mice, Nogo-A/Bf/f HFD n = 7 mice and Nogo-A/BECKO HFD n = 5 mice). I, J PE-induced vasoconstriction (Nogo-A/Bf/f SD n = 8 mice, Nogo-A/BECKO SD n = 10 mice, Nogo-A/Bf/f HFD n = 4 mice and Nogo-A/BECKO HFD n = 5 mice). K, L Myogenic tone (Nogo-A/Bf/f SD n = 8 mice, Nogo-A/Bf/f HFD n = 4 mice, Nogo-A/BECKO SD n = 5 mice and Nogo-A/BECKO HFD n = 7 mice). M, N Ach-mediated vasodilation (Nogo-A/Bf/f SD n = 11 mice, Nogo-A/BECKO SD n = 10 mice, Nogo-A/Bf/f HFD n = 7 mice and Nogo-A/BECKO HFD n = 8 mice); O, P flow-induced vasodilation (Nogo-A/Bf/f SD n = 7 mice, Nogo-A/BECKO SD n = 8 mice, Nogo-A/Bf/f HFD n = 6 mice and Nogo-A/BECKO HFD n = 8 mice), and Q flow-induced vasodilation after myriocin treatment (1 mg/kg, i.v., o.n.), (n = 3 mice per group). LC–MS/MS quantification of R total and S specific Cer, C16:0-dihydroceramide (dhC16:0-Cer), dihydrosphingosine (dhSph), sphingosine (Sph), sphingosine-1-phosphate (S1P), and T total and U specific SM in EC FACS-sorted from heart and lung of Nogo-A/B-deficient and WT mice fed with HFD for 6 months. Data are expressed relative to WT HFD (Nogo-A/Bf/f HFD n = 18 mice and Nogo-A/BECKO HFD n = 17 mice). V Heatmap of gene for the SPT complex, inflammation, and lipid transport in FACS-sorted EC from heart and lung, *indicates significant differences (Nogo-A/Bf/f HFD n = 4 mice and Nogo-A/BECKO HFD n = 4 mice). Data were expressed as mean ± SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. Statistical analysis was performed with two-way ANOVA with Sidak multiple comparisons test (B, C, D, E, I–Q), one-way ANOVA (F–H) unpaired t-test two-tailed (R–U). Source data are provided as a Source Data file.
Discussion
Multiple studies support the ‘endothelial-centric’ role in the pathogenesis of cardiometabolic diseases, a growing health problem among the population in USA and worldwide5,36. Thus, understanding the mechanisms underlying endothelial dysfunction in cardiometabolic diseases is imperative for the development of appropriate therapeutic strategies to cure these life-treating conditions.
Multiple evidence has linked dysregulation of sphingolipid metabolism, particularly ceramide, to obesity-associated metabolic dysfunctions, including insulin resistance, glucose intolerance, diabetes, and steatohepatitis37,38. While robust clinical evidence identified plasma ceramides as independent predictors of major cardiovascular events31,39,40,41, how sphingolipid metabolism changes in the vasculature and contribute to vascular diseases remains poorly understood.
Although the endothelium contributes to plasma sphingolipids16,42, it is not the only source. Indeed, plasma sphingolipids, including ceramides, are rather a surrogate originating from different sources (i.e. liver, fat, bone marrow cells, etc.). Our study investigates how HFD changes sphingolipid metabolism and levels specifically in the vasculature and does not argue against the role of ceramide as predictor of major cardiovascular event.
It has been postulated that ceramide accrual in the EC and arteries causes dysfunction and contributes to hypertension and atherosclerosis, especially when cardiometabolic disorders coexist. Findings from this study are paradigm shift because demonstrate that ceramide deficit and not the accrual in EC and resistance arteries causes dysfunction in obesity.
Exposing in vitro EC to high glucose and/or palmitate boosts ceramide biosynthesis and accrual19,20,21, and data from our lab in HUVEC (Supplementary Fig. 8) support these findings. On the contrary, EC from obese mice present a deficit of ceramides (Fig. 1H, I), resulting from the suppression of sphingolipid de novo biosynthesis by Nogo-B and ORMDLs, overexpressed in HFD (Fig. 1D–F). RNAseq data of EC isolated from obese and diabetic mice showed a significant increase in Rtn4 (the gene for Nogo-B) and Ormdl2,3, suggesting a transcriptional upregulation of these inhibitors in line with protein levels. However, we did not find a correlation between the changes in the mRNA for Sptlc1 and Sptlc2 (Fig. 1G) and their protein levels, which were unchanged. This is not surprising since we have observed the same outcome when assessing the Sptlc1 and Sptlc2 expression in cardiomyocytes isolated from pressure-overloaded hearts6.
The suppression of sphingolipids correlated with impaired endothelial dysfunction and hypertension. Interestingly, the loss of Nogo-B preserved endothelial sphingolipid levels in mesenteric arteries of HFD mice (Fig. 5H–K), and significantly improved endothelial dysfunction and hypertension compared to obese WT mice (Fig. 5B–G and Fig. 4). S1P and ceramides are metabolically interchangeable and both regulates EC functions4. In HFD, both ceramides and S1P are suppressed in EC (Fig. 1H, I). We have shown that genetic repression of sphingolipid de novo biosynthesis impacts both ceramide and S1P production16. Furthermore, by using different genetic approaches targeting Nogo-B11, S1P transporter Spns28, S1P19, and S1P39 we have shown that local S1P-S1P1 signaling regulates blood flow and pressure8,9,11, and it suppresses inflammation43. The loss of Nogo-B improved the vasodilation in response to flow of MA from obese mice, and W146, inhibitor of S1P1, abolished this effect, suggesting that S1P signaling was mainly accountable for the improved endothelial-mediated vasorelaxation to flow.
Findings from this study also demonstrated that endothelial-derived S1P downregulates hepatic gluconeogenesis via S1P1. Growing evidence support an active role of the endothelium in influencing the metabolism of surrounding cells/tissues by secreting different factors, such as NO, growth factors and enzymes44. For instance, endothelial-secreted PDGF-CC induces white adipose tissue browning45, while endothelial lipase plays an important role in fatty acid metabolism, particularly in hearts exposed to hemodynamic stress46. In addition to hypertension, genetic deletion of eNOS also leads to insulin resistance and hyperlipidemia, underlying the important role of NO in metabolism47,48. Thus, as potent activator of eNOS via S1P1,3, S1P exerts also beneficial metabolic and cardiovascular functions via NO. In obese mice Nogo-B-mediated suppression of sphingolipid de novo biosynthesis in the endothelium contributes to enhance hepatic gluconeogenesis and worsen diabetes (Figs. 3 and 6). Different studies have shown that modulation of S1P signaling by liver over-expression of sphingosine kinase 1 (Sphk1)28 and Sphk229,49 have beneficial effects on hepatic glucose metabolism, supporting the role of S1P as negative regulator of hepatic gluconeogenesis. Our data show that S1P of non-parenchymal origin, specifically from the endothelium, reduces hepatic glucose production via S1P1 signaling (Figs. 2O and 6E). In vivo data in Nogo-A/BECKO obese mice corroborated the in vitro data showing that both exogenous S1P and co-culture of EC and hepatocytes suppressed hepatic gluconeogenesis. As Gi-coupled receptor, S1P1 activation decreases cellular levels of cAMP50 and activates Akt signaling51, both inhibitors of hepatic gluconeogenesis52,53.
The suppression of ceramides by Nogo-B-mediated inhibition of SPT also contributes to endothelial dysfunction. Recently, we reported that endothelial deletion of Sptlc2 subunit of SPT, impairing sphingolipid de novo biosynthesis, suppressed both ceramides and S1P. Although the activation of eNOS by S1P was blunted, the decrease of ceramides boosted eNOS phosphorylation and NO production16, most likely by keeping PP2A in a low activation state, in agreement with the study of Zhang and colleagues showing that elevation of ceramides leads to eNOS dephosphorylation via PP2A19. Despite the beneficial effects of NO, the suppression of ceramides impaired endothelial-mediated vasodilation in response to flow and signal transduction16, both resulting in dysfunction. This is not surprising since ceramides can also modulate biophysical properties of lipid rafts,54,55, specialized signaling platforms.
Considering that SPT activity is enhanced by substrate availability56 and inflammatory mediators, it is counterintuitive to assume the suppression of sphingolipid biosynthesis EC exposed to an environment overwhelmed of glucose, inflammatory mediators, and lipids, including palmitate which is the main lipid used as substrate by SPT. It is possible that sphingolipid downregulation in the endothelium of obese and diabetic mice is an attempt of compensation, although the dysregulation of this pathway both in excess and deficit results in impaired cellular functions.
The deletion of Nogo-B restores sphingolipid biosynthesis and decreased pro-inflammatory gene expression as shown by the RNAseq data (Fig. 6V). It is known that obesity triggers chronic inflammation that contributes to atherosclerosis57.
These findings causally implicate the suppression of endothelial sphingolipid signaling by Nogo-B in the onset of an inflammatory and atherosclerotic phenotype of the endothelium in cardiometabolic diseases. The decrease of sphingolipid signaling by Nogo-B can impact the transcriptional signature by different means. For instance, the decrease of ceramide was reported to activate the NF-kB pathway via retrotranslocation of PKCα/β from the membrane58, while the ablation of endothelial S1P1 signaling enhances the expression of adhesion molecules, such as VCAM1 and ICAM1, NF-kB activation and atherosclerosis43. Altogether, these findings suggest that S1P and ceramides needs to be maintained within a physiological range to preserve endothelial homeostasis.
Ceramide was found elevated in the aorta of obese mice19, hence was postulated that ceramide accrual in resistance arteries contributed to increased vasocontraction and hypertension. Our findings demonstrated that resistance arteries (i.e. MA) of obese mice showed a significant decrease of ceramides and SM, in line with Nogo-B upregulation in MA (Fig. 1L), which correlated with hypercontractility (Fig. 5D–G). Conversely, in mice lacking Nogo-B, vascular ceramides were preserved and vasoconstriction in response to PE, AngII, and intravascular pressure were significantly decreased. These functional data were corroborated by decreased MLC and MYPT1 phosphorylation in MA in response to AngII (Fig. 5L–N). Of note, the loss of Nogo-B specifically in EC per se contributes to reduce vascular contractility (Fig. 6I–L).
Some limitations of our study include the measure of sphingolipids and gene expression in EC isolated from heart and lung, comprising different vascular beds, and the use of only male mice.
In conclusion, our study investigates in vivo mechanisms underlying the dysregulation of sphingolipid metabolism and signaling in the vasculature and its pathological implications in cardiometabolic disease.
The novelty of our study is the demonstration in vivo that ceramide and S1P are suppressed in the endothelium of mice fed a HFD, and the deletion of the inhibitor Nogo-B restores sphingolipid levels. This restoration of sphingolipid levels is associated with improved endothelial and metabolic functions in obese mice (Fig. 7). These results demonstrate that endothelial ceramide suppression underlies cardiometabolic diseases. In this regard, Manzo et al. recently demonstrated that ceramide accumulation did not contribute to endothelial dysfunction in coronary atherosclerosis induced by hemodynamic stress in ApoE−/− mice59. While in both models of cardiometabolic diseases ceramide levels do not increase, the underlying mechanisms are dramatically different. In the endothelium of obese mice, the de novo biosynthesis is suppressed. In contrast, in ApoE−/− mice under hemodynamic stress, the de novo biosynthesis is upregulated together with increased ceramide degradation, resulting in the rewiring of sphingolipid metabolism towards S1P, which is protective. The deletion of endothelial Nogo-B sustains this effect in long-term protecting the mice from coronary atherosclerosis. The changes of sphingolipid de novo pathway in the endothelium of mice fed a HFD are diametrically opposite to the changes occurring in endothelium of coronary atherosclerosis, suggesting that the regulation of this metabolic pathway is disease dependent. These findings define a framework for the designing of therapeutic approaches to treat different cardiometabolic conditions.

(Created in BioRender. Di Lorenzo, A. (2025) https://BioRender.com/i50g226).
Methods
Animals
Nogo-A/B-deficient mice were generated as described by Kim et al.60. These mice have been backcrossed with C57BL/6 for more than 8 generations. WT (C57BL/6) littermate mice were used as controls. Mice Nogo-A/Bf/f were generated as previously described11. Mice lacking Nogo-A/B specifically in endothelial cells (Nogo-A/BECKO) were obtained by crossing Nogo-A/Bf/f mice with transgenic mice in which the VE-cadherin promoter drives the expression of Cre (VE-Cad-CreERT2), selectively deleting the loxP-flanked (‘floxed’) region of Rtn4 in endothelial cells, as previously reported11. Male mice at 6–7 weeks of age were provided standard chow diet (SD) or HFD (60% fat; #D12492i, Research diet) for 6 months. The research animal facility housing the mice has a controlled 12-h light/12-h dark light cycle and temperature of 65–75 °F (~18–23 °C) with 40–60% humidity. Certified veterinarians in laboratory animal medicine and veterinary pathology oversee all aspects of animal care. All the studies were performed according to protocols approved by the Weill Cornell Institutional Animal Care and Use Committee.
Endothelial cells FACS sorting
At 6 months of HFD or SD, endothelial cells were FACS-sorted from heart and lung. Briefly, organs were digested in PBS containing Collagenase Type I 2 mg/mL (Alfa Aesar, cat# J62406), Dispase 0.5 U/mL (StemCell, cat# 07913) and DNaseI 100μg/mL (Roche, # 10104159001) Cardiomyocytes were depleted by using a cell strainer of 40 mm and the non-cardiomyocyte cell suspension was stained with Alexa 488-labeled Isolectin B4 (Invitrogen, #I21411) and antibodies against CD31 for positive selection (Biolegend, #102407), and CD45 and Ter119 for negative selection (Biolegend, #103125 and #116223 respectively). After FACS-sorting, EC were collected by centrifugation and stored at −80 °C for downstream analysis (western blot, sphingolipid measurement, RNAseq).
Western blot analysis
WB were performed as previously reported11,16. Briefly, endothelial cells or mesenteric arteries were homogenized in RIPA buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Sodium Deoxycholate, 1% NP-40, 0.1% SDS, 5 mM NaF, 1 mM Na3VO4, protease and phosphatase inhibitors) and analyzed with sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting. The following primary antibodies were used: Nogo-B (R&D, #AF6034), Ormdl3 (Millipore, #ABN417), SPTLC1 (BD Biosciences, #611305), SPTLC2 (Abclonal, ##A11716), phospho-S19-MLC, MLC, phospho-T696-MYPT1 (Cell Signaling Technology, #3671, #3672 and 5163 respectively), MYPT1 (sc-514261, SantaCruz Biotechnology) β-Actin (ThermoFisher Scientific, #AM4302). Following exposure to HRP-conjugated secondary antibodies, reactive bands were visualized with ECL reagents (#RPN2236, Amersham), and band density analyzed with ImageJ.
Sphingolipid analysis by LC/MS/MS
FACS-sorted EC from heart and lung of mice were homogenized in RIPA buffer. Mesenteric arteries were dissected and cleaned from surrounding fat tissues, snap frozen on dry ice and stored at −80 °C. Analysis for sphingolipid contents was performed by LC/MS/MS at the Lipidomics Shared Resource at the Medical University of South Carolina.
RNA sequencing
RNA was extracted from FACS-sorted EC using TRIzol reagent protocol (ThermoFisher Scientific, #15596026). RNA-seq was performed by RNA core of Weill Cornell Medicine (New York, NY). Total RNA integrity was evaluated with the Agilent Bioanalyzer 2100 (Agilent Biotechnologies, Santa Clara, CA), and only samples with an RNA integrity number (RIN) > 9.0 were further processed for RNA-seq. All RNA-seq libraries were prepared using 100 ng of total RNA with the TruSeq Stranded Total RNA sample preparation kit (ribosomal RNA depletion and stranded RNA-Seq) with Ribo-Zero according to the manufacturer’s specified protocol (Illumina, San Diego, CA). Samples were multiplexed and sequenced across multiple lanes of a HiSeq 4000 Sequencing System (Illumina) using 50-base paired-end reads to achieve a minimum depth of 30 million aligned read-pairs. Data were analyzed by the Englander Institute for Precision Medicine of Weill Cornell Medicine (New York, NY).
Glucose and insulin tolerance tests
Nogo-A/B-deficient and WT male mice at 6–7 weeks of age were fed with standard diet (SD) or a high-fat diet (HFD; D12492i; Research Diets, Inc.) for 6 months. At the end of this treatment, mice were assessed for glucose and insulin tolerance tests. by using glucose (G7021; Sigma-Aldrich) and insulin For glucose tolerance test (GTT), mice were fasted o.n. and injected with a glucose(G7021; Sigma-Aldrich) solution i.p. (1.5 g/kg BW) and blood glucose was measured at the indicated times in the figures, by using the Bayer Contour glucose meter. For the insulin tolerance test61, mice were fasted for 4.5 h and received an injection of insulin (Actrapid; Novo Nordisk; 0.75 U/kg BW for SD-fed mice; and 1.5U/kg body weight for HFD-fed mice). Blood glucose was measured at the indicated times in the figures, using the Bayer Contour glucose meter.
Real-time PCR
Total RNA from liver and brown adipose tissue was extracted according to the TRIzol reagent protocol (ThermoFisher Scientific, #15596026). For liver Maxima First Strand cDNA Synthesis Kit (ThermoFisher Scientific, #K1641) was used for the reverse transcription of 100 ng of RNA. For RT-PCR PowerUp™ SYBR™ Green Master Mix (ThemoFisher Scientific, #A25779) and Applied Biosystems 7500 Fast RT-PCR system were used. Primers set were: G6Pase (5′-AGGAAGGATGGAGGAAGGAA-3′ and 5′-TGGAACCAGATGGGAAAGAG-3′) and 18S (5′-TTCCGATAACGAACGAGACTCT-3′ and 5′-TGGCTGAACGCCACTTGTC-3′). Gene of interest relative mRNA expression was calculated with the 2(−ΔΔCt) method, using 18S as housekeeping62. For RT brown adipose tissues High-Capacity cDNA Reverse transcription Kit (Cat# 4368814, Thermo Fisher Scientific) was used for the reverse transcription of 100 ng of RNA. Real-time PCR with diluted cDNAs was performed using the LightCycler 480 (Roche) and Taqman Gene Expression Assay primers (Thermo Fisher Scientific) in triplicates. Gene expression of the target genes was normalized to β-actin. The calculations of average Cp values, SDs, and resulting expression ratios for each target gene were performed using the Roche LightCycler 480 software. The following Primers were used: Ucp1, Mm01244861_m1; Dio2, Mm00515664_m1; β-Actin, Mm02619580_g1 (ThermoFisher Scientific, #4331182).
Measurement of circulating hormones
Serum insulin was measured following the manufacturer’s protocol (CrystalChem, #90080) before and after glucose injection (0.75 U/kg for SD-fed mice and 1.5U/Kg for HFD-fed mice, 20’). Serum leptin was measured following the manufacturer’s protocol (Millipore, #EZML-82K).
Glucose, insulin, and pyruvate tolerance tests
At the end of the 6 months of diet, mice were assessed for glucose and insulin tolerance. Glucose tolerance test (GTT) was performed in mice fasted o.n. After determination of basal blood glucose levels, mice were injected i.p. with glucose 1.5 g/kg (G7021; Sigma-Aldrich), and blood glucose was measured at the indicated times by using the Bayer Contour glucometer. Insulin tolerance test61 was performed in mice were fasted for 4.5 h. After determination of basal blood glucose levels, mice were injected i.p with insulin (0.75 U/kg for SD fed mice; 1.5U/Kg for HFD-fed mice. Actrapid; Novo Nordisk). Blood glucose was measured at the indicated times by using the Bayer Contour glucose meter. Pyruvate Tolerance Test (PTT) was performed in mice fasted o.n. After determination of basal blood glucose concentrations, mice were injected i.p. with sodium pyrivate 1.5 g/kg (P4562, Sigma), and blood glucose was measured at the indicated times by using the Bayer Contour glucometer.
Radiotelemetry BP measurements
Systolic, diastolic, and mean blood pressure were measured in conscious Nogo-A/B-deficient and control mice fed with SD or HFD up to 6 months using Data Sciences International (DSI) implantable radiotelemetry transmitters8,16. Anesthetized (isoflurane, to effect) mice were implanted with carotid artery catheters advanced to the aortic arch and radiotelemeter implants (model HD-X10) inserted in a subcutaneous pocket on the back. After ca. 12 days of recovery, BP was monitored continuously, with values reported every 5 s, for three consecutive days.
Vascular reactivity studies
After 6 months of SD or HFD, Nogo-A/B-deficient and control mice were sacrificed and second order mesenteric arteries (MA) were harvested, cleaned from adhering tissue and mounted on glass micropipettes in a pressure myograph chamber (Danish MyoTechnology, Aarhus, Denmark), as previously described11,16, by keeping the orientation of the vessel in relation to the flow in vivo. MA were maintained viable in Krebs solution (mmol/L: NaCl 118, KCl 4.7, MgCl2 1.2, KH2PO4 1.2, CaCl2 2.5, NaHCO3 25, and glucose 10.1), warmed (37 °C) and oxygenated (95% O2 and 5% CO2)11,16. After 15 min of equilibration at 80 mmHg, MA were pre-constricted with PE (1 μM) and a cumulative concentration-response curves of Ach (0.1 nM to 30 μM) were performed to evaluate the endothelial function. Endothelial-dependent vasodilation is response to stepwise increase in flow was evaluated as previously described11,16. In same settings, MA were incubated with W146 (300 nM, 45 min) followed by flow-induced vasodilation. The concentration-response curves of phenylephrine (PE, 1 nM to 30 µM), angiotensin-II (AngII, 1nM-30µM) were also performed. In another set of experiments, myogenic response was performed by stepwise increase of intraluminal pressure (20–45–70–95–120 mmHg) and each pressure level was maintained for 5 min. At the end of the experiment, a myogenic curve was performed in Ca2+-free Krebs buffer supplemented with 1 mol/L EGTA. Myogenic tone (%) was expressed as [(D1 − D2)/D1]·100, where D1 is the passive diameter in Ca2+-free Krebs buffer and D2 is the active diameter in complete Krebs buffer, at the same intraluminal pressure.
Immunofluorescence staining
Pancreas from Nogo-A/B deficient and control mice. on chow and high-fat diets, were harvested, washed in PBS and rapidly fixed in 4% PFA o.n. at 4 °C. After 24 h of incubation in 30% sucrose, at 4 °C, pancreas was cryostated in OCT. Sections of 8 μm were co-stained for Nogo-A/B (1:100, R&D) and insulin (1:100, BD Biosciences) or glucagone (1:100, XXX) overnight at 4 °C. Following PBS washing steps, pancreas sections were incubated with secondary antibodies, Alexa 594-labeled anti-rat and streptavidin-Alexa 488 in PBS for 1 h at room temperature. Nuclei were stained with DAPI. Immunofluorescence images of the tissues were captured with Zeiss 2012 epifluorescent microscope.
Immunohistochemistry
For immunostaining of Nogo-B, liver cross sections were incubated overnight at 4 °C with anti-Nogo-A/B antibody (1:100; #AF6034, R&D Systems), followed by secondary anti-sheep Biotin-SP (1:200, 713-065-003; Jackson Immuno Research). The staining was developed with diaminobenzene, and sections were counterstained with Mayer’s hematoxylin, dehydrated, and coverslipped with mounting medium. Light microscopic images were acquired using a Zeiss Axio Observer.Z1 microscope.
Primary hepatocyte isolation and experimental procedure
Primary hepatocytes were harvested for the measurement of glucose production as previously described63. Briefly, 7–11-week-old mice were anesthetized using a mixture of ketamine (100 mg/kg body weight; Webster Veterinary) and xylazine (10 mg/kg body weight; Webster Veterinary). Livers were perfused with 12 mL of liver perfusion media for 4 min (Invitrogen) followed by 30 mL of liver digestion media for 8 min (Invitrogen) at 37 °C. Digested hepatocytes were released into 50 mL ice-cold hepatocyte wash media (Invitrogen) and filtered through 70-micron filter (BD Biosciences). Dead cells were removed using Percoll gradient (Sigma-Aldrich), and remaining healthy hepatocytes were washed with 50 mL wash buffer and resuspended in ice-cold M199 media (Invitrogen) containing 10% FBS, penicillin (100 U/mL) and streptomycin (100 μg/mL). Isolated hepatocytes were plated at the density of 5 × 105 cells per well on collagen-coated plates. Alternatively, isolated hepatocytes were plated directly on pre-isolated mouse EC. Hepatocytes were allowed to attach for 6 h prior to serum-starvation overnight. The next morning, hepatocytes were washed twice with phosphate-buffered saline (PBS; Gibco) followed by incubation for 6 h in glucose- and phenol red-free Dulbecco’s modified Eagle’s medium (DMEM) (15 mM HEPES [pH 7.4] and 3.7 g/liter NaHCO3) supplemented with 20 mM sodium pyruvate (Sigma) and 20 mM lactate (Sigma) as gluconeogenic substrates. The influence of S1P and W146 was determined throughout this 6 h incubation. Glucose concentrations were measured enzymatically using the glucose (HK) assay kit (Sigma) according to the manufacturer’s protocol by determining the absorbance of the cell culture media at 340 nm using Sprectramax i3x Multi-Mode Microplate Reader (Molecular Devices) and normalized to cellular protein content.
Statistical analysis
Data were expressed as mean ± SEM. One- or two-way ANOVA were used for all statistical analyses except where Student’s t-test analysis was used. Differences were considered statistically significant when P < 0.05. All the tests were two-sided. GraphPad Prism software (version 6.0, GraphPad Software, San Diego, CA) was used for all the statistical analysis.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data generated or analyzed during this study are included in this published article and its Supplementary Information. Source data are provided with this paper. All RNA-seq data generated in this study have been deposited in GEO with the accession code GSE278555 and its corresponding hyperlink https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE278555 Source data are provided with this paper.
References
Powell-Wiley, T. M. et al. Obesity and cardiovascular disease: a scientific statement from the American Heart Association. Circulation 143, e984–e1010 (2021).
Hadi, H. A. & Suwaidi, J. A. Endothelial dysfunction in diabetes mellitus. Vasc. Health Risk Manag. 3, 853–876 (2007).
Al Suwaidi, J., Higano, S. T., Holmes, D. R. Jr., Lennon, R. & Lerman, A. Obesity is independently associated with coronary endothelial dysfunction in patients with normal or mildly diseased coronary arteries. J. Am. Coll. Cardiol. 37, 1523–1528 (2001).
Sasset, L. & Di Lorenzo, A. Sphingolipid metabolism and signaling in endothelial cell functions. Adv. Exp. Med. Biol. 1372, 87–117 (2022).
Graupera, M. & Claret, M. Endothelial cells: new players in obesity and related metabolic disorders. Trends Endocrinol. Metab. 29, 781–794 (2018).
Sasset, L. et al. Nogo-A reduces ceramide de novo biosynthesis to protect from heart failure. Cardiovasc. Res. 119, 506–519 (2022).
Sasset, L., Zhang, Y., Dunn, T. M. & Di Lorenzo, A. Sphingolipid de novo biosynthesis: a rheostat of cardiovascular homeostasis. Trends Endocrinol. Metab. 27, 807–819 (2016).
Del Gaudio, I. et al. Endothelial Spns2 and ApoM regulation of vascular tone and hypertension via sphingosine-1-phosphate. J. Am. Heart Assoc. 10, e021261 (2021).
Cantalupo, A. et al. S1PR1 (Sphingosine-1-Phosphate Receptor 1) signaling regulates blood flow and pressure. Hypertension 70, 426–434 (2017).
Venkataraman, K. et al. Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circ. Res. 102, 669–676 (2008).
Cantalupo, A. et al. Nogo-B regulates endothelial sphingolipid homeostasis to control vascular function and blood pressure. Nat. Med. 21, 1028–1037 (2015).
Obinata, H. et al. Individual variation of human S1P(1) coding sequence leads to heterogeneity in receptor function and drug interactions. J. Lipid Res. 55, 2665–2675 (2014).
Shendre, A. et al. Local ancestry and clinical cardiovascular events among african americans from the atherosclerosis risk in communities study. J. Am. Heart Assoc. 6, e004739 (2017).
Domarkiene, I. et al. RTN4 and FBXL17 genes are associated with coronary heart disease in genome-wide association analysis of lithuanian families. Balk. J. Med. Genet. 16, 17–22 (2013).
Pinto, S. N., Silva, L. C., Futerman, A. H. & Prieto, M. Effect of ceramide structure on membrane biophysical properties: the role of acyl chain length and unsaturation. Biochim. Biophys. Acta 1808, 2753–2760 (2011).
Cantalupo, A. et al. Endothelial sphingolipid de novo synthesis controls blood pressure by regulating signal transduction and NO via ceramide. Hypertension 75, 1279–1288 (2020).
Chalfant, C. E. et al. Long chain ceramides activate protein phosphatase-1 and protein phosphatase-2A. Activation is stereospecific and regulated by phosphatidic acid. J. Biol. Chem. 274, 20313–20317 (1999).
Dobrowsky, R. T., Kamibayashi, C., Mumby, M. C. & Hannun, Y. A. Ceramide activates heterotrimeric protein phosphatase 2A. J. Biol. Chem. 268, 15523–15530 (1993).
Zhang, Q. J. et al. Ceramide mediates vascular dysfunction in diet-induced obesity by PP2A-mediated dephosphorylation of the eNOS-Akt complex. Diabetes 61, 1848–1859 (2012).
Bharath, L. P. et al. Ceramide-initiated protein phosphatase 2A activation contributes to arterial dysfunction in vivo. Diabetes 64, 3914–3926 (2015).
Mehra, V. C. et al. Ceramide-activated phosphatase mediates fatty acid-induced endothelial VEGF resistance and impaired angiogenesis. Am. J. Pathol. 184, 1562–1576 (2014).
Li, P. L. & Zhang, Y. Cross talk between ceramide and redox signaling: implications for endothelial dysfunction and renal disease. Handb. Exp. Pharmacol. 216, 171–197 (2013).
Breslow, D. K. et al. Orm family proteins mediate sphingolipid homeostasis. Nature 463, 1048–1053 (2010).
Pechlivani, N. & Ajjan, R. A. Thrombosis and vascular inflammation in diabetes: mechanisms and potential therapeutic targets. Front. Cardiovasc. Med. 5, 1 (2018).
Stratmann, B. & Tschoepe, D. Sweet heart - contributions of metabolism in the development of heart failure in diabetes mellitus. Exp. Clin. Endocrinol. Diabetes 116, S40–S45 (2008).
Bonal, C. B. et al. Nogo-A downregulation improves insulin secretion in mice. Diabetes 62, 1443–1452 (2013).
Zhang, D. et al. Reticulon 4B (Nogo-B) is a novel regulator of hepatic fibrosis. Hepatology 53, 1306–1315 (2011).
Ma, M. M. et al. Sphingosine kinase 1 participates in insulin signalling and regulates glucose metabolism and homeostasis in KK/Ay diabetic mice. Diabetologia 50, 891–900 (2007).
Aji, G. et al. Regulation of hepatic insulin signaling and glucose homeostasis by sphingosine kinase 2. Proc. Natl Acad. Sci. USA 117, 24434–24442 (2020).
Turpin, S. M. et al. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab. 20, 678–686 (2014).
Laaksonen, R. et al. Plasma ceramides predict cardiovascular death in patients with stable coronary artery disease and acute coronary syndromes beyond LDL-cholesterol. Eur. Heart J. 37, 1967–1976 (2016).
Touyz, R. M. et al. Vascular smooth muscle contraction in hypertension. Cardiovasc. Res. 114, 529–539 (2018).
Kim, Y. R. et al. Ablation of ceramide synthase 2 exacerbates dextran sodium sulphate-induced colitis in mice due to increased intestinal permeability. J. Cell Mol. Med. 21, 3565–3578 (2017).
Chen, Y. H. et al. Molecular cloning of cDNA encoding the 110 kDa and 21 kDa regulatory subunits of smooth muscle protein phosphatase 1M. FEBS Lett. 356, 51–55 (1994).
GrandPre, T., Nakamura, F., Vartanian, T. & Strittmatter, S. M. Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature 403, 439–444 (2000).
Bierhansl, L., Conradi, L. C., Treps, L., Dewerchin, M. & Carmeliet, P. Central role of metabolism in endothelial cell function and vascular disease. Physiology 32, 126–140 (2017).
Chaurasia, B. & Summers, S. A. Ceramides - lipotoxic inducers of metabolic disorders. Trends Endocrinol. Metab. 26, 538–550 (2015).
Summers, S. A., Chaurasia, B. & Holland, W. L. Metabolic Messengers: ceramides. Nat. Metab. 1, 1051–1058 (2019).
de Carvalho, L. P. et al. Plasma ceramides as prognostic biomarkers and their arterial and myocardial tissue correlates in acute myocardial infarction. JACC Basic. Transl. Sci. 3, 163–175 (2018).
Havulinna, A. S. et al. Circulating ceramides predict cardiovascular outcomes in the population-based FINRISK 2002 cohort. Arterioscler. Thromb. Vasc. Biol. 36, 2424–2430 (2016).
Peterson, L. R. et al. Ceramide remodeling and risk of cardiovascular events and mortality. J. Am. Heart Assoc. 7, e007931 (2018).
Kuo, A. et al. Murine endothelial serine palmitoyltransferase 1 (SPTLC1) is required for vascular development and systemic sphingolipid homeostasis. Elife 11, e78861 (2022).
Galvani, S. et al. HDL-bound sphingosine 1-phosphate acts as a biased agonist for the endothelial cell receptor S1P1 to limit vascular inflammation. Sci. Signal. 8, ra79 (2015).
Pi, X., Xie, L. & Patterson, C. Emerging roles of vascular endothelium in metabolic homeostasis. Circ. Res. 123, 477–494 (2018).
Seki, T. et al. Endothelial PDGF-CC regulates angiogenesis-dependent thermogenesis in beige fat. Nat. Commun. 7, 12152 (2016).
Nakajima, H. et al. Endothelial lipase modulates pressure overload-induced heart failure through alternative pathway for fatty acid uptake. Hypertension 61, 1002–1007 (2013).
Duplain, H. et al. Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase. Circulation 104, 342–345 (2001).
Cook, S. et al. Partial gene deletion of endothelial nitric oxide synthase predisposes to exaggerated high-fat diet-induced insulin resistance and arterial hypertension. Diabetes 53, 2067–2072 (2004).
Lee, S. Y. et al. Activation of sphingosine kinase 2 by endoplasmic reticulum stress ameliorates hepatic steatosis and insulin resistance in mice. Hepatology 62, 135–146 (2015).
Means, C. K., Miyamoto, S., Chun, J. & Brown, J. H. S1P1 receptor localization confers selectivity for Gi-mediated cAMP and contractile responses. J. Biol. Chem. 283, 11954–11963 (2008).
Morales-Ruiz, M. et al. Sphingosine 1-phosphate activates Akt, nitric oxide production, and chemotaxis through a Gi protein/phosphoinositide 3-kinase pathway in endothelial cells. J. Biol. Chem. 276, 19672–19677 (2001).
Li, X., Monks, B., Ge, Q. & Birnbaum, M. J. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature 447, 1012–1016 (2007).
Wang, Y. et al. Inositol-1,4,5-trisphosphate receptor regulates hepatic gluconeogenesis in fasting and diabetes. Nature 485, 128–132 (2012).
Kolesnick, R. N., Goni, F. M. & Alonso, A. Compartmentalization of ceramide signaling: physical foundations and biological effects. J. Cell Physiol. 184, 285–300 (2000).
Simons, K. & Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 1, 31–39 (2000).
Rutti, M. F., Richard, S., Penno, A., von Eckardstein, A. & Hornemann, T. An improved method to determine serine palmitoyltransferase activity. J. Lipid Res. 50, 1237–1244 (2009).
Henning, R. J. Obesity and obesity-induced inflammatory disease contribute to atherosclerosis: a review of the pathophysiology and treatment of obesity. Am. J. Cardiovasc. Dis. 11, 504–529 (2021).
Signorelli, P., Luberto, C. & Hannun, Y. A. Ceramide inhibition of NF-kappaB activation involves reverse translocation of classical protein kinase C (PKC) isoenzymes: requirement for kinase activity and carboxyl-terminal phosphorylation of PKC for the ceramide response. FASEB J. 15, 2401–2414 (2001).
Manzo, O. L. et al. Rewiring endothelial sphingolipid metabolism to favor S1P over ceramide protects from coronary atherosclerosis. Circ. Res. 134, 990–1005 (2024).
Kim, J. E., Li, S., GrandPre, T., Qiu, D. & Strittmatter, S. M. Axon regeneration in young adult mice lacking Nogo-A/B. Neuron 38, 187–199 (2003).
Gangoda, L. et al. Inhibition of cathepsin proteases attenuates migration and sensitizes aggressive N-Myc amplified human neuroblastoma cells to doxorubicin. Oncotarget 6, 11175–11190 (2015).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 (2001).
Steensels, S. et al. Acyl-Coenzyme A thioesterase 9 traffics mitochondrial short-chain fatty acids toward de novo lipogenesis and glucose production in the liver. Hepatology 72, 857–872 (2020).
Acknowledgements
This work was supported by the National Institutes of Health grants R01 HL126913 and R01 HL152195 to A.D.L.; R01 DK136079 and DK131717 to S.D.; R01 DK129576 to B.E.
Author information
Authors and Affiliations
Contributions
L.R. initiated the project and carried out experiments. O.L.M. carried out experiments, analyzed data, and wrote the manuscript. J.S. and S.D. carried out experiments and analysis relative to energy expenditure. L.R. and I.D.G. performed vascular function studies on mesenteric arteries. R.B. and O.E. analyzed and deposited the RNAseq data. A.M. executed blood pressure measurement by telemetry. S. P. performed FACS analysis. M. B. contributed to interpretation of data and manuscript revision. D. F. carried out the histology analysis. B. E. responsible for hepatocyte experiments. L.S. performed FACS-sorting endothelial cell from murine heart and lung, western blot analysis, sphingolipid measurement analysis. V. D. M. Sphingolipid measurements in HUVEC exposed to high glucose and palmitate. A.D.L conceived the idea, supervised the project, and wrote the manuscript. All the authors were involved in revising the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Camilla Wenceslau and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rubinelli, L., Manzo, O.L., Sungho, J. et al. Suppression of endothelial ceramide de novo biosynthesis by Nogo-B contributes to cardiometabolic diseases. Nat Commun 16, 1968 (2025). https://doi.org/10.1038/s41467-025-56869-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56869-9
